近日,中国科学技术大学核科学技术学院马骏教授团队基于辐射化学原理,提出并实现了一种针对全氟和多氟烷基物质(PFAS)的彻底矿化新路径。该研究聚焦于三氟乙酸——碳链最短、最难降解的PFAS代表物,设计出由氧化与还原协同驱动的自由基串联反应机制,在室温、无外加催化剂的条件下,实现了污染物的近乎完全矿化。相关成果以“The O•–/electron tandem path for complete mineralization of trifluoroacetate and perfluorocarboxylic acids”为题,发表于《自然·水》(Nature Water)期刊。
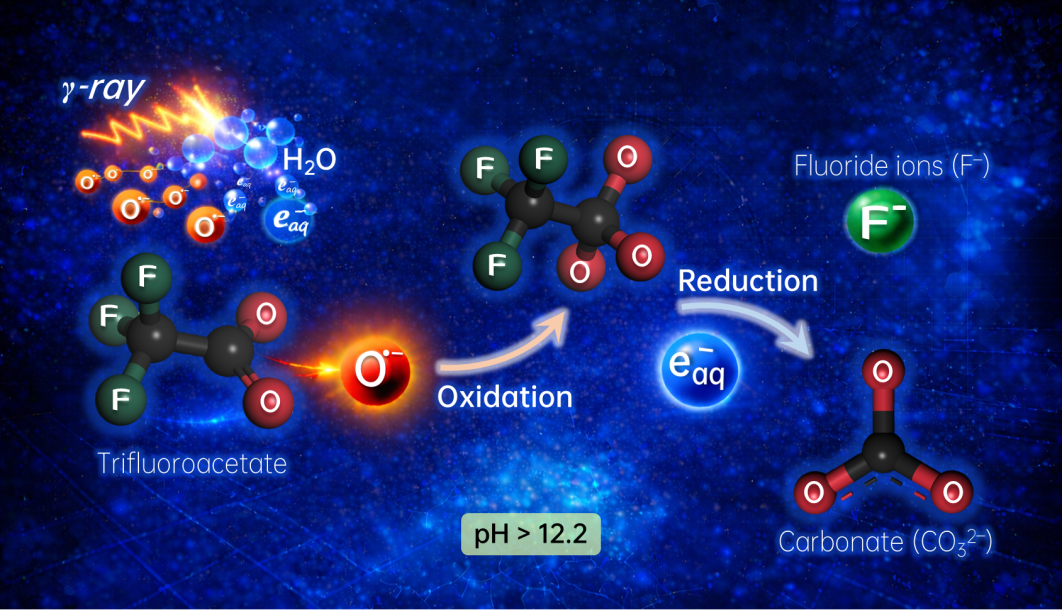
PFAS因化学稳定性强、应用广泛,被大量用于制药、农用化学品、表面处理及高分子材料等领域。然而,其C–F键极高的稳定性也导致这类化合物在环境中极难降解,易于长期残留并形成持续污染,故被称为“永久污染物”。在各类PFAS中,三氟乙酸的降解尤为困难。作为碳链最短的PFAS,三氟乙酸具有更强的环境迁移性与持久性,同时也是多数长链PFAS的最终降解产物,被视为实现PFAS完全降解的“最后一公里”。相较于部分长链PFAS,三氟乙酸对传统光化学、电化学及热化学降解路径表现出更强的惰性,现有技术往往难以实现高效脱氟与彻底矿化。因此,如何突破其反应惰性、建立新的活化降解机制,已成为PFAS污染控制领域的关键科学难题。
受地球早期海洋中天然辐射过程可清除有害物质的启发,研究团队利用高能辐射在水中产生的高活性物种,提出了由氧负离子自由基(O•–)和水合电子(eaq−)共同介导的氧化–还原协同降解机制。通过脉冲辐解等时间分辨研究手段,团队系统揭示了高能辐射诱导的自由基串联降解三氟乙酸的完整过程。研究发现,O•–能够优先氧化活化三氟乙酸,开辟了传统羟基自由基(•OH)难以触达的反应通道,并以较高反应速率生成关键中间体。该中间体进一步与水合电子反应,最终实现C–F键断裂,并完全矿化为氟离子和碳酸根。实验结果显示,在此协同路径下,三氟乙酸可在六小时内达到接近100%的矿化率。
该研究不仅成功突破了三氟乙酸这类超短链PFAS的降解瓶颈,更从机制层面更新了对PFAS活化路径的传统认知。长期以来,学界普遍认为PFAS难以通过氧化途径实现深度转化,而本研究证明,在特定活性物种参与下,氧化步骤不仅可行,还能在后续还原过程中发挥关键活化作用,从而构建出一条高效的串联降解路径。
除三氟乙酸外,研究团队进一步验证了该策略对多种全氟羧酸及全卤代羧酸均具有良好的降解效果,显示出较强的普适性。与此同时,研究还完成了工业电子束辐照条件下的预中试验证,获得了0.27 mol·L⁻¹·h⁻¹的脱氟速率,表明该方法与现有高通量水处理技术具备良好的衔接基础,为未来工程化应用提供了重要支撑,展现出高能辐射技术在水体污染治理中的广阔前景。
法国国家科学研究中心终身研究员姜志文(中国科大1620校友)为论文第一作者,法国国家科学研究中心Mehran Mostafavi教授与中国科学技术大学核科学技术学院马骏教授为共同通讯作者。该研究工作得到了国家自然科学基金等项目的支持。
论文链接:The O•−/electron tandem path for complete mineralization of trifluoroacetate and perfluorocarboxylic acids. Nature Water. 2026, https://www.nature.com/articles/s44221-026-00632-x